
The Lancet Summit:
Presymptomatic Prevention and Treatment of Neurodegenerative Diseases
14 - 16 December 2021 | Virtual and On-Demand
The biological paths that lead to Alzheimer’s disease and other neurodegenerative disorders start many years before clinical symptoms arise. In research settings, diagnostic criteria for these diseases now involve the use of biomarkers that reveal such pathophysiological changes.
Researchers are identifying and refining genetic, radiological, and CSF and blood biomarkers. At the same time, the effects on these biomarkers of therapies that could halt or delay neurodegeneration are being tested in clinical trials. These advances could ultimately enable Alzheimer’s disease and other neurodegenerative disorders to be prevented at preclinical stage.
Landmark biomarker and prevention studies, and hypothetical models of preclinical neurodegenerative diseases have been reported in The Lancet and The Lancet Neurology over the past decade. Reflecting our commitment to support this crucial area of neurological research, this Summit will provide a timely update on the latest advances and challenges in this area, with an emphasis on prevention.
The Summit will also provide a forum for discussion of the ethical and regulatory issues that these advances will bring about in research settings and clinical practice.
The programme will cover these topics:
- Genetics, cellular pathways, and neuronal vulnerability
- Biomarkers and early diagnosis
- Prevention through therapeutics
- Ethical and regulatory considerations
Participate live from your desktop or mobile device to:
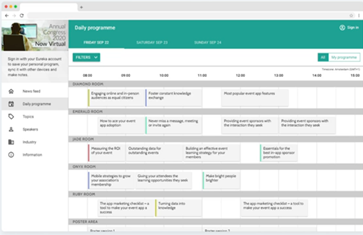
Deepen your knowledge: Live-stream a personalised schedule from the entire programme to match your research interests.
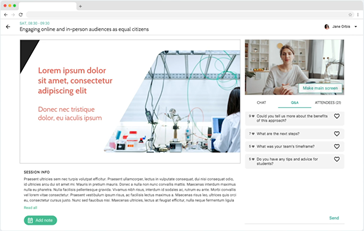
Engage with our inspirational speakers and other attendees during the live-streamed sessions: Ask questions and participate in live chat and polls.
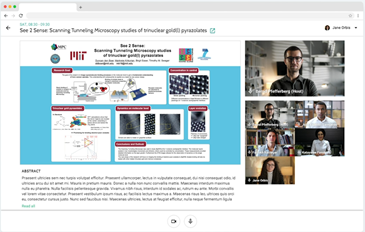
Review the latest research in the ePoster hall and discuss directly with the author in the live poster sessions.
Plus:
- Network with your peers, meet new contacts, and renew relationships: Connect with other attendees during the live event to chat and arrange one-to-one meetings.
- Communicate your research to a wider, more-inclusive audience: our lower fees, online delivery and post-event on-demand access will make the event more accessible to all.
- Visit the live exhibition booths to meet industry partners, chat with exhibitors, and learn more about their products
- Enjoy more flexibility with on-demand access to recorded sessions for 6 months after the event*. For multi-stream events, watch one stream live and catch-up on what you missed after the event.
* On-demand access includes recordings of talks and slides, poster pitches and posters from authors that have agreed for post event distribution: the organisers cannot guarantee on-demand access to all presentations.
#LancetSummit
-
Adam Boxer, USA
Alexandra Durr, France
Ammar Al-Chalabi, UK
David Klenerman, UK
Feng Zhang, USA
Henrik Zetterberg, UK
Holly Kordasiewicz, USA
John Hardy, UK
Jonathan Rohrer, UK
Karen Duff, UK
Lorenzo Guizzaro, Netherlands
Nick Fox, UK
Patricia Saidon, Argentina
Rachelle Doody, Switzerland
Reisa Sperling, USA
Robert Klitzman, USA
Roy Yaari, USA
Sarah Tabrizi, UK
Sonia Vallabh and Eric Minikel, USA
Sophie Leggat, UK
Timothy Miller, USA
Virginia Lee, USA
- Elena Becker-Barroso, Editor, The Lancet Neurology, UK
- Helen Frankish, Executive Editor, The Lancet, UK
- Nick Fox, UK Dementia Research Institute, UK
- John Hardy, UK Dementia Research Institute, UK
- Jonathan Rohrer, Dementia Research Centre, UCL, London, UK
- Sarah Tabrizi, UK Dementia Research Institute, London, UK




